
阿茲海默症不只是「類澱粉斑塊」或「tau纏結」的單一路徑,而是一場跨越蛋白質錯誤摺疊與神經發炎的複雜交響。這篇文章以病理解剖與靈長類模型為基礎,整合了amyloid、tau與炎症反應三者間的互動網絡,揭示它們如何在不同病程階段彼此牽動。
重點提問
Q1:如何整合阿茲海默症研究中,關於 Tau 與 Amyloid 病理「何者為始」的長期矛盾觀點?
Q2:本研究特別強調「老化獼猴模型」的價值,它如何彌補人類死後腦研究的關鍵知識缺口?
Q3:除了形成神經纖維纏結,早期的「可溶性磷酸化 Tau」在細胞內扮演了哪些具體的致病角色?
Q4:新興的體液生物標記 pT217Tau 為何與「Amyloid-PET」的關聯性,甚至比「Tau-PET」更強?
Q5:鈣離子失衡(Calcium Dysregulation)如何在阿茲海默症病理中,扮演串連遺傳、發炎、Tau 與 Amyloid 的核心樞紐角色?
Q6:如何重新定義傳統的「Amyloid 假說」,提出一個更符合散發性 AD 病程的模型?
An integrated view of the relationships between amyloid, tau, and inflammatory pathophysiology in Alzheimer's disease
文獻出處
背景
- 現存知識與矛盾:阿茲海默症(AD)的診斷需要 Amyloid 斑塊與 Tau 神經纖維纏結(NFTs)的證據,但關於何者為病理的始作俑者,學界存在長期爭論 。
Amyloid 假說:受家族性 AD 遺傳學支持,認為 Aβ42 的產生是起始事件,進而驅動 Tau 病理與神經退化 。
神經病理學觀點:對散發性 AD(sAD)的研究顯示,Tau 的病理內含物(inclusions)比 Amyloid 的沉積早出現約十年,對 Amyloid 假說構成挑戰 。
本文目的:整合多元研究方法,包括 流體生物標記 與 老化獼猴模型,以重新建構 AD 發病早期的完整圖像。
Tau and Amyloid的細胞基礎生物學
Tau
生理功能:Tau 蛋白在正常生理狀態下,主要功能是調節並穩定微管結構,同時也參與細胞核內 DNA 的調控 。它廣泛分佈於大腦皮質神經元的樹突與軸突中,對於維持廣闊樹突結構的神經元尤其重要 。
病理轉變過程:
過度磷酸化 (Hyperphosphorylation):病理起始於Proline-rich region (PRR)的過度磷酸化,這使得帶正電的 PRR 轉為帶負電,導致其構型改變 。
從微管脫離:結構型改變使 Tau 的微管結合區(MTBR)從微管上脫離,損壞微管穩定性 。磷酸化是循序發生的,早期位點包括 T231、T181、T217,晚期位點則如 pT205(AT8 抗體辨識位點) 。
可溶性 pTau 階段 (Soluble pTau):
此為最早期的病理狀態,pTau 仍是可溶的,且理論上可被磷酸酶去磷酸化 。
在死後 15 分鐘內即被迅速去磷酸化,因此病理解剖難以捕捉。
可溶性 pTau 具有毒性,會干擾細胞健康,並可在神經元間傳播 。
與流體生物標記 pT217Tau 高度相關,成為早期檢測窗口。
聚集與纖維化 (Aggregation and Fibrillation):可溶性 pTau 在進一步的轉譯後修飾(如乙醯化、泛素化)後,開始聚集成半可溶的「前纏結(pretangle)」 。最終,它們堆疊形成不可溶、嗜銀性的神經纖維纏結(NFTs)
病程演變序列:
可溶性 tau → 聚集於微管形成 pretangle → 均勻分布於胞體樹突(AT8 標記)→ 疊合形成不溶性纖維 (NFTs)。
晚期:NFT 僅剩「MTBR 核心纖維」,失去 AT8 免疫反應。
影像學限制:tau-PET 僅能偵測中間階段,無法捕捉最早期或最晚期 tau。
Amyloid (Aβ)
- 生理功能:澱粉樣前驅蛋白(APP)是一種膜蛋白,其正常功能仍在研究中,但已知在腦部發育與突觸可塑性中扮演重要角色 。
APP 結構與生成路徑:
APP 為跨膜糖蛋白,含小型細胞內段 (AICD) 與 Aβ 區域。
在 內涵體:β- 與 γ-分泌酶切割 → 生成 Aβ42 與 AICD。
在 細胞膜:α-分泌酶切割 → 阻止 Aβ42 生成。
APOE ε4 改變 APP 內涵體內定位,加速 Aβ42 生成。
聚集與毒性機制:
Aβ42 從單體 → 寡聚體 → 原纖維 → 纖維化斑塊。
寡聚體/原纖維 為主要毒性來源:
結合 prion 蛋白 → 異常 mGluR5 活化 → 突觸喪失、tau 磷酸化。
增加細胞內鈣離子,誘發鈣失衡。
與鈣離子穩態的關聯:
γ-secretase 複合體(包含 Presenilin 1/2)除了切割 APP,也調節 SERCA 幫浦的活性,此幫浦負責將鈣離子泵入內質網儲存 。
家族性 AD 的 PSEN1/PS2 基因突變會損害 SERCA 功能,導致胞漿鈣離子濃度過高,啟動細胞毒性反應
惡性循環:
Aβ42 → tau 磷酸化與突觸喪失。
Tau 聚集與內涵體捕捉 → 促進 Aβ42 生成。
高鈣激活 calpain-2、cdk5、GSK3β → 進一步惡化 tau/Aβ 病理。
發炎與 APOE ε4 加劇此循環。
區域性差異:
類澱粉沉積範圍廣泛,與 tau 病理侷限於內嗅皮質形成對比。
PET 影像顯示在聯合皮質特別明顯,但內嗅皮質相對少見。
誰是始作俑者?視角決定觀點
來自人類大腦神經病理學的觀點
優勢:是 AD 診斷的黃金標準,具有細胞層級的空間解析度 。
劣勢:只能在單一時間點進行橫斷面分析,缺乏縱向資料。且因死後延遲(PMI),會遺失最早期、不穩定的可溶性 pTau 病理 。
核心觀點:Tau 病理早於 Aβ 沉積約十年 。
Tau 病理始於特定皮質下核區,隨後進入內嗅皮質(entorhinal cortex, ERC)第二層的投射神經元 。
病理從樹突的遠端開始,逐漸向近端及細胞本體發展,最後才影響軸突 。
Tau 病理的擴散遵循一個可預測的模式(Braak stages I-VI),從內側顳葉擴散至整個新皮質 。
認知功能障礙的嚴重程度與 Tau 纏結及突觸的喪失密切相關,而非 Aβ 斑塊的負荷量 。
爭議:此文反對原發性年齡相關 Tau 病變(PART)是一種獨立疾病的假說,認為其 Tau 蛋白摺疊結構與 AD 相同,應視為 AD 的臨床前期 。
來自正子斷層掃描 (PET) 影像的觀點
優勢:可在活體進行縱向追蹤,並同時評估多種病理標記(Amyloid, Tau, 突觸密度等)。並能與 MRI 與 FDG-PET 結合,提供結構與功能互補資訊。
劣勢:目前的示蹤劑靈敏度與空間解析度有限,只能偵測到已纖維化的 Aβ 或 Tau,無法看到早期的可溶性病理 。特別是 Tau-PET,通常要到神經病理學 NFT stage IV 之後才能穩定偵測到訊號 。
核心觀點:Amyloid-PET 訊號的出現早於 Tau-PET 訊號 。
在活體影像中,皮質的纖維狀 Amyloid 沉積先出現,之後才在內側顳葉偵測到纖維狀 Tau 的訊號 。
Amyloid-PET 影像的出現時點大約對應到死後病理分期的 Aβ phase 3,顯示 PET 偵測的時程落後於實際的分子病理變化 。
FDG-PET 顯示,與症狀相關的腦部葡萄糖代謝下降,發生在 Amyloid-PET 陽性之後 。
SV2A-PET 顯示,突觸的喪失與 Tau 病理的關聯性較高,而與 Amyloid 沉積無關,此結果與神經病理學一致 。
來自體顯性遺傳 AD (ADAD) 的遺傳學觀點
優勢:提供了明確的致病機制,直接指向類澱粉蛋白病理的核心角色 。
劣勢:這些罕見的顯性遺傳病例可能無法完全推廣至臨床上佔絕大多數的晚發性散發性 AD(sAD)。
核心觀點:Aβ42 病理是疾病進程的始動因子 。
- APP 基因重複,以及 PSEN1、PSEN2 基因突變(γ-secretase 的組成部分)均會導致 AD
- 此觀點催生了「類澱粉假說」:Aβ42 是「扳機」,而 Tau 是殺死細胞的「子彈」。
抗 Aβ42 抗體療法可降低 amyloid-PET 與 tau-PET,延緩病程,但女性與 APOE ε4 攜帶者效果較差。
部分攜帶 PSEN1 突變者若合併 APOE-ε3 或 reelin 保護性突變,儘管有大量 Aβ,卻缺乏 tau 病理與失智 → Aβ42 不是唯一必要條件。
來自晚發散發性 AD (sAD) 的遺傳學觀點
優勢:研究結果與絕大多數 AD 病例相關,並指向發炎與脂質代謝等多重機制 。
劣勢:除了 APOE 基因型外,其他相關基因位點眾多、效應小且複雜,難以形成單一簡潔的致病理論 。
核心觀點:發炎反應是驅動疾病病理的關鍵因素 。
APOEε4 是 sAD 最強的遺傳風險因子,會增加 Aβ42 聚集並降低其清除效率 。
- GWAS 確認超過 130 個易感位點,多與發炎、內涵體運輸、鈣失衡相關。
代表性基因:
BIN1:促進 tau 播散、增加鈣通道。
TREM2:調控小膠質細胞對 Aβ42 清除能力。
PICALM:參與網格蛋白介導的內吞、自噬。
SORL1:突變造成內涵體功能障礙。
DNA 甲基化研究顯示 BIN1 與 Aβ42、SORL1 與 pTau 水準相關。
來自 sAD 大腦轉錄體學與蛋白質體學的觀點
優勢:提供無偏見的全景式分析,有助於發現新的致病機制與細胞特異性弱點 。
劣勢:數據量龐大;可能遺失重要的轉譯後修飾(如磷酸化tau);且難以區分因果關係與次級代償反應 。
核心觀點:目前尚無統一結論,但研究結果普遍指向發炎反應、粒線體功能失調、突觸失能、內涵體-溶酶體功能障礙及鈣信號異常等 。
- 新皮質的 Tau 蛋白含量比Aβ42 含量更能預測認知能力的急劇下降,與神經病理學觀點一致 。
- 蛋白質體學分析顯示,sAD 與家族性 AD 在分子層面的特徵驚人地相似 。
- 常見蛋白變化:GFAP、CamKK2、細胞骨架相關蛋白。
早期失去 SST GABAergic 神經元、reelin 表達中間神經元,可能導致網絡不穩。
常見蛋白變化:GFAP、CamKK2、細胞骨架相關蛋白。
來自體液生物標記的觀點
優勢:提供一個早期、微創、且具成本效益的方式來追蹤疾病病理,有望實現臨床前診斷 。
劣勢:是腦內實際病生理過程的間接指標;體液中的 Tau 生物標記主要是細胞釋放的片段,可能無法完全反映神經元內的完整病理狀態
核心觀點:Aβ42 與 pT217Tau 是最早出現的 AD 體液生物標記 。
Aβ42:腦脊髓液(CSF)中 Aβ42 的濃度下降,被認為是 Aβ 在腦中沉積為斑塊的反映,與 Amyloid-PET 訊號的出現同步 。
pTau:血漿與 CSF 中的 pT217Tau 極早期就會升高,甚至早於 Tau-PET 訊號的出現,並與未來疾病的發生高度相關 。pT217Tau 的早期升高與 Amyloid-PET 訊號的出現有極強的關聯性 。
MTBR-tau243:這是一種來自 Tau 微管結合區的片段,其在 CSF 中的濃度升高與晚期的不可溶 Tau 纏結及 Tau-PET 訊號密切相關,可作為疾病進展的後期標記 。
優勢:老化獼猴是研究 sAD 早期病理的理想模型 。
- 有 高度發展的聯絡皮層(association cortex),與人類相似。
其 谷氨酸能神經元展現出 放大版的鈣訊號,是病理高風險區域。
基因特徵與人類接近:如 3R/4R tau 混合型,APOE ε4 同合子。
血漿 pT217Tau 隨年齡增加,與人類生物標記趨勢相同。
自然發展 類澱粉斑塊與 tau 病理,進展順序與人類相符:
ERC layer II → NFT(超過 30 歲的極老猴子)。
NFT 的結構為 paired helical filaments(PHFs),與人類相同。
並伴隨 明顯的記憶缺損。
劣勢:
獼猴壽命 20–40 年,在實驗室中 真正的極老齡猴極為稀少(COVID 疫苗研究消耗了大量猴隻)。
病理雖然質性相似,但量化程度較低:最老猴子大約僅達到人類 NFT stage III 的水平。
核心觀點:早期的可溶性 pTau 與 Aβ 病理,和鈣離子失衡密切相關,三者相互作用形成惡性循環 。
- 早期可溶性 Aβ42 與 pTau → 鈣失衡
包括 calbindin 的喪失(一種保護性鈣結合蛋白)。
惡性循環(vicious cycles):
鈣失衡 ↔ tau 磷酸化 ↔ 類澱粉過量產生 ↔ 自噬性退化。
pT217Tau:
具高度神經毒性。
能在突觸間 傳播與「感染」其他神經元。
與 APP 裂解增加相關(內吞小泡被「困住」→ 促進 Aβ42 生成)。
血漿 pT217Tau 與腦內 amyloid-PET 信號高度相關。
- 早期可溶性 Aβ42 與 pTau → 鈣失衡
疫電子顯微鏡(ImmunoEM)發現:
可溶性 pTau(尤其是 pT217Tau)
聚集於 樹突微管上。
干擾 內體運輸 → 阻塞 endosome → 增加 Aβ42 產生。
引發 自噬小泡累積與粒線體功能異常 → 神經毒性。
pTau + Aβ42 + Ca2+ 失衡 → 惡性循環,持續加劇病理。
隨著 tau 進一步纖維化(AT8 標記):
樹突內的 **自噬性退化(autophagic degeneration)**明顯。
破壞細胞合成 Aβ42 的能力 → 解釋 ERC 為何澱粉樣沉積量少。
突觸傳播:
pTau 可在 興奮性突觸間傳遞 → 「感染」整個聯絡迴路。
小部分可溶性 pTau 會 暴露到細胞外液 → 被 CSF 與血漿檢測到(流體生物標記來源)。
整合人類與獼猴數據及未來研究方向
整合研究結果
可溶性 Aβ42 與可溶性 tau (pTau) 幾乎同時出現,是 AD 病理最早期的事件。
但由於 Aβ42 會主動排出至細胞外,容易被 CSF 或血漿捕捉,再加上 PET 成像解析度不足(只能偵測較晚期的纖維化 tau),因此在臨床上會「誤以為」是類澱粉先行。
獼猴模型揭示了 人腦病理解剖難以看見的早期 tau 事件 ——這正好解釋了為什麼血液或腦脊髓液裡的 tau 生物標記物(如 pT217Tau)會「提前很久」升高,而 tau-PET 卻遲遲無法偵測。
病理進程的連續光譜
tau 的早期可溶性階段 → 提供了血液中的 tau 訊號,也促進 Aβ42 的生成。
進入纖維化 tau 階段 → 才能被死後病理解剖或 tau-PET 偵測到,但這其實已經是比較後期的病理。
進一步膜破壞與自噬退化 → tau 的 MTBR 片段(如 Tau243)釋出,可在晚期疾病的體液中測得。
未來研究與臨床啟示
更靈敏的 PET 影像劑:能捕捉 tau 的早期可溶性型態與不同類型的發炎反應(不只針對小膠細胞,還包含神經元層級)。
更精確的體液生物標記物:能區分 tau 的不同聚集階段,及早預測病程。
發炎與鈣離子失衡作為早期關鍵驅動因子:臨床上或許應及早針對這些機制進行干預。
獼猴模型的價值:因其天然帶有 APOE ε4、發炎狀態、與人類高度相似的皮質結構,極適合用來測試 早期抗發炎策略。
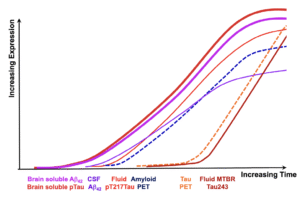
最早期的分子事件(腦內變化):
腦內可溶性 pTau (Brain soluble pTau) 和 腦內可溶性 Aβ42 (Brain soluble Aβ42):圖中最先開始上升的是這兩條曲線 。是同時開始在腦中累積的 。這些是最初的分子病變,但無法被目前的臨床影像技術直接偵測。
早期可偵測的生物標記(體液與 PET 影像):
體液 pT217Tau (Fluid pT217Tau):緊隨腦內可溶性 pTau 的出現,體液(腦脊髓液和血漿)中的 pT217Tau 濃度開始上升 。這表示腦內早期的可溶性 Tau 病理已開始釋出至體液中,成為我們能偵測到的第一個 Tau 病理訊號 。
腦脊髓液 Aβ42 (CSF Aβ42):這是一條特殊的向下曲線 。幾乎與體液 pT217Tau 上升的同時,腦脊髓液中的 Aβ42 濃度開始下降。這是因為腦內的 Aβ42 開始聚集成斑塊並沉積,導致其在腦脊髓液中的濃度降低,這是一個反向的關鍵指標 。
類澱粉蛋白 PET (Amyloid PET):在 CSF Aβ42 下降後不久,Amyloid PET 的訊號開始上升 。這代表腦中已累積了足夠多的纖維化類澱粉蛋白斑塊,達到了能被 PET 影像偵測到的閾值。
中晚期的生物標記:
Tau PET:這條曲線的上升時間點遠遠晚於 Amyloid PET 和體液 pT217Tau 。這反映了目前 Tau PET 配體的技術限制——它們對早期的可溶性 Tau 不敏感,只能偵測到疾病進展到較後期、已形成大量纖維化神經纖維纏結(NFTs)的 Tau 病理 。
體液 MTBR-Tau243 (Fluid MTBR-Tau243):這條曲線的上升趨勢與 Tau PET 非常接近 。文獻指出,MTBR-Tau243 是一個專門反映不可溶的 Tau 纏結病理的體液標記,通常在神經元退化較嚴重的晚期階段才會釋出 。
AD 病理與治療策略的關聯性
- 抗類澱粉治療(Anti-amyloid therapies)
背景:AD 治療的第一波突破,源自長期以來對「類澱粉假說」的聚焦。
已獲 FDA 完全核准的藥物:
Lecanemab (Leqembi)【靶點:可溶性 Aβ 原纖維(protofibrils)】
優勢:瞄準早期病程的主要毒性物種。
臨床:證實能延緩疾病進展。
Donanemab (Kisunla)【靶點:不溶性、N-端截斷的 pyroglutamate Aβ(存在於成熟斑塊)】
優勢:針對斑塊核心病理。
臨床:同樣證實可延緩疾病進展。
限制與風險:
副作用:ARIA(Amyloid-Related Imaging Abnormalities)
ARIA-E:水腫或滲出。
ARIA-H:腦微出血、大出血或鐵質沉積。
APOE ε4 攜帶者(尤其同合子):
更容易出現 ARIA。
風險更高,且可能病程中伴隨更強的炎症驅動。
臨床意涵:雖然抗類澱粉抗體療法開啟了治療新時代,但安全性與療效仍需提升。
抗 tau 治療(Anti-tau strategies)
現況:
多種策略已嘗試,但至今 尚無成功案例。
N-端 tau 抗體:未能減緩臨床惡化,也未能降低 PET 影像中的 tau 聚集。
新一代策略:
瞄準 MTBR(Microtubule Binding Region)
優勢:只有 tau 異常磷酸化並脫離微管時才會暴露,代表已進入病理性狀態。
潛力:或能 更精準鎖定致病 tau,降低副作用。
展望:期望能比抗類澱粉療法更具療效且副作用更少。
間接干預路徑(Indirect approaches)
核心理念:不是直接清除 Aβ42 或 tau,而是 抑制其產生或促進清除,特別針對 炎症與氧化壓力。
科學基礎:
炎症與氧化壓力 → 鈣離子失衡 → tau/Aβ 異常產生。
糖尿病 → 風險因子:
誘導氧化壓力/鈣失衡。
增加 GSK3β 活性 → tau 磷酸化 & 類澱粉生成。
治療探索:
GLP-1 受體促進劑(GLP-1 receptor agonists)
機轉:改善葡萄糖代謝、減少炎症與鈣失衡。
臨床證據:隨機臨床試驗綜合分析 → 顯著降低「全因性失智症」風險。
代表性藥物:Semaglutide(大型臨床試驗進行中,NCT04777396 & NCT04777409)。
鼻腔胰島素(intranasal insulin)
初步結果顯示改善潛力。
本研究與既有文獻在觀點上有哪些一致性與分歧?
- 一致性:
承認 AD 的多因性:Aβ、Tau 蛋白、神經發炎及鈣離子失衡等都是病理網絡中的關鍵節點
Tau 病理與臨床症狀的強相關性:Tau 神經纖維纏結(NFTs)的數量和分佈,比 Aβ 斑塊的負荷更能準確預測患者的認知功能下降程度 。
肯定神經發炎的核心角色
- 新觀點
支持並賦予 Tau 更主動的角色:傳統上被視為 Aβ 下游受害者的 Tau 蛋白,在本文的模型中被賦予了更早、更主動的致病能力。
將發炎與鈣離子假說置於核心:本文實質上是將「發炎假說」與「鈣離子假說」置於其整合模型的最上游。它清晰地論述了發炎信號如何導致鈣離子失衡,而鈣離子失衡又是如何成為一個「共同的下游通路」,透過活化 Calpain-2、GSK3β、Cdk5 等酵素,同時驅動了 Aβ 的生成和 Tau 的過度磷酸化 。這使得發炎與鈣離子不再是伴隨現象,而是驅動整個惡性循環的引擎。
個人想法
- 此文章整合老化獼猴模型和人體大腦的病理研究 ,嘗試解釋了為何血漿 pT217Tau 能如此早期出現並預測疾病,值得有後續的研究去證實。
- 雖然獼猴模型是理解早期病理的一個好工具 ,但文章中也承認其病理嚴重程度終究不如晚期人類患者 。將模型中觀察到的細胞機制(如內涵體堵塞)的影響力,完全等同於人類複雜的散發性 AD 病程,這個推論鏈仍需更多直接的人類證據來證實。
- 這個循環模型也解釋了為何單純靶向類澱粉蛋白的藥物療效有限 。它強力地暗示,未來的治療必須是「組合療法」,例如結合抗類澱粉、抗發炎及穩定鈣離子通道的多重策略
- 文章相當肯定地將原發性年齡相關 Tau 病變(PART)歸類為 AD 的臨床前期 。儘管其引用的證據有力,但 PART 的獨立性在學界仍有持續的討論 。
重點問答
Q1:如何整合阿茲海默症研究中,關於 Tau 與 Amyloid 病理「何者為始」的長期矛盾觀點?
傳統神經病理學觀察到,在散發性 AD(sAD)患者的死後腦組織中,Tau 蛋白的病理變化(early pretangle stages a–c, 1a, 1b, and NFT stages I–III i)比 Amyloid-β(Aβ)斑塊的沉積早 。然而,活體 PET 影像學與家族性 AD 的遺傳學研究則強烈支持 Amyloid 假說,認為 Aβ 的產生是啟動 Tau 病理的關鍵驅動因子 。
此文提出一個整合性觀點:這並非一個簡單的線性因果關係,而是Aβ42、磷酸化 Tau(pTau),以及發炎反應/鈣離子失衡三者間形成的「前饋循環(feedforward cycles)」 。研究認為,各領域的矛盾源於其方法學上的盲點,例如死後腦組織研究無法偵測到早期可溶性的 pTau,而現行 PET 技術對早期 Tau 病理的靈敏度不足 。透過整合新興的體液生物標記與獼猴模型數據,本文試圖描繪一個更完整的早期病理圖像。
Q2:本研究特別強調「老化獼猴模型」的價值,它如何彌補人類死後腦研究的關鍵知識缺口?
老化獼猴模型之所以關鍵,在於它能以極短的死後間隔或透過灌流固定的方式取得腦組織,從而保存了在人類死後腦組織中極易降解的「可溶性磷酸化 Tau(soluble pTau)」 。研究指出,人類腦組織在死後 15 分鐘內,可溶性 pTau 就會被快速去磷酸化,導致這種最早期的病理狀態在傳統神經病理學研究中幾乎是「隱形」的 。然而,這正是 Tau 病理的起始與具備毒性的關鍵階段。獼猴模型的高解析度電子顯微鏡研究,首次清晰地展示了可溶性 pTau 在神經元樹突中聚集、干擾內涵體運輸(endosomal trafficking),並可能促進 Aβ42 的生成 。這項證據不僅解釋了為何新興的體液生物標記(如 pT217Tau)能比 PET 影像更早偵測到 Tau 病理,也為 Tau 與 Amyloid 之間的早期互動提供了直接的細胞生物學基礎。
Q3:除了形成神經纖維纏結,早期的「可溶性磷酸化 Tau」在細胞內扮演了哪些具體的致病角色?
在形成不可溶的纖維纏結之前,可溶性 pTau 本身就具有顯著的細胞毒性。透過獼猴模型的高解析度影像,研究觀察到可溶性 pTau(包含 pT217Tau)會在樹突的微管上聚集 。這種聚集會干擾微管的穩定性與正常的內涵體運輸功能,如同造成了細胞內的「交通堵塞」 。這種堵塞會使含有 APP 蛋白的內涵體被「困住」,延長其在內涵體內被 β-secretase 和 γ-secretase 切割的時間,從而增加了致病性 Aβ42 的產量 。此外,這種早期 Tau 病理狀態與自噬小泡的累積和粒線體異常有關,最終導致樹突的退化 。同時,可溶性 pTau 也能在神經元之間的突觸進行傳播,將病理擴散至相連的腦區 。
Q4:新興的體液生物標記 pT217Tau 為何與「Amyloid-PET」的關聯性,甚至比「Tau-PET」更強?
這是一個看似矛盾卻極具啟發性的觀察。此研究提出的整合模型解釋了此現象。
血漿 pT217Tau 反映的是非常早期的「可溶性」Tau 病理,而目前的 Tau-PET 配體僅能偵測到晚期已形成纖維束的「不可溶」Tau 。獼猴模型的證據顯示,這種早期的可溶性 pTau 會在樹突中聚集並干擾內涵體運輸,進而增加神經元內部 Aβ42 的產出 。因此,當大腦中開始出現可溶性 pTau 病理時,會伴隨著 Aβ42 的加速生成與後續的斑塊沉積。體液中的 pT217Tau 濃度升高,實際上是與大腦中 Aβ 加速累積的過程同步發生。這解釋了為何 pT217Tau 能成為預測大腦中 Amyloid 沉積(由 Amyloid-PET 測得)的強力指標 。相對地,當 Tau-PET 訊號變得顯著時,通常已是疾病較晚期的階段,此時 Tau 病理已大規模纖維化。
Q5:鈣離子失衡(Calcium Dysregulation)如何在阿茲海默症病理中,扮演串連遺傳、發炎、Tau 與 Amyloid 的核心樞紐角色?
此文將鈣離子失衡定位為驅動 sAD 病理的共同通路。多種上游因素均指向這個核心機制。
首先,家族性 AD 的 PSEN1/PSEN2 基因突變會損害 SERCA 幫浦功能,導致胞漿鈣離子濃度過高 。其次,sAD 的主要遺傳風險因子(如 APOEε4)與多種發炎相關基因,都會加劇鈣離子穩態的失調 。當胞漿內鈣離子濃度持續過高,會活化下游的酵素 Calpain-2 。被活化的 Calpain-2 會啟動一系列致病反應:它會切割並「去抑制」GSK3β 與 Cdk5 這兩種激酶,使其大量磷酸化 Tau 蛋白 ;同時,Cdk5 也會增加 APP 的切割,促進 Aβ42 生成 。Aβ42 本身及其片段(AICD)又會反過來促進鈣離子從內質網釋放,形成惡性循環 。因此,鈣離子失衡不僅是單一的病理現象,而是一個整合了遺傳易感性、發炎壓力,並同時驅動 Tau 與 Amyloid 病理的中心節點。
Q6:如何重新定義傳統的「Amyloid 假說」,提出一個更符合散發性 AD 病程的模型?
傳統的 Amyloid 假說(Amyloid Cascade Hypothesis)提出一個線性的、階層式的病程:Aβ 異常累積是起始事件,隨後觸發 Tau 病理、神經元死亡與認知功能下降 。然而,此模型難以解釋為何在 sAD 中 Tau 病理會比 Aβ 斑塊更早出現 。
此文提出的「前饋循環」模型(Feedforward Cycle Model)則認為,在 sAD 中,上游的驅動因子更可能是老化相關的發炎反應與鈣離子失衡 。這些因素可獨立或協同地引發早期的可溶性 pTau 病理與 Aβ42 的產生。重要的是,這三者(pTau、 Aβ42、鈣離子/發炎)之間會互相增強、彼此放大,形成一個自我持續的惡性循環 。例如,pTau 透過干擾內涵體來增加Aβ42 的生成;而 Aβ42 則會加劇 Tau 的磷酸化與鈣離子失衡。這個模型更動態、非線性,更能解釋 sAD 的複雜性,並為為何針對單一靶點的藥物效果有限提供了理論基礎。