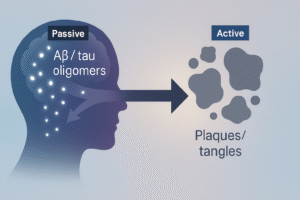
這篇來自劍橋大學團隊的文章,對阿茲海默症的傳統病理模型提出了根本性的修正。文章的核心論點是,我們所熟知的 Aβ 斑塊與 Tau 蛋白纏結,可能並非疾病的主要驅動者,而是細胞在面對真正具高度毒性的「可溶性寡聚體」時,所採取的積極防禦措施。作者們整合了生物物理學與細胞生物學的證據,構建了「多階段動態平衡假說」,主張 AD 的本質是蛋白質生成與清除之間的失衡,而非單純的沉積。
重點提問
Q1:傳統的類澱粉蛋白假說將 Aβ 斑塊與 Tau 纏結視為核心病理,本文的核心論點如何對此提出修正?
Q2:鑑於腦中 Aβ 單體濃度遠低於自發聚集的門檻,本文如何解釋大規模 Aβ 斑塊形成?
Q3:神經纖維纏結 (NFTs) 的數量與認知能力下降高度相關,但本文對其在細胞層面的角色提出了什麼不同的解釋?
Q4:如果斑塊和纏結並非病理的根本原因,這套「動態平衡假說」認為阿茲海默症的觸發點是什麼?
Q5:此假說如何為 Lecanemab 與 Donanemab 這兩種 Aβ 抗體藥物看似不同的作用機制提供統一的解釋框架?
Q6:若此假說成立,對於阿茲海默症未來的診斷策略與研究方向有何意義?
Small-diffusible aggregates, plaques, tangles, and dynamic equilibria: Untangling Alzheimer's disease
文獻出處
背景
歷史沿革與里程碑
早期紀錄:古希臘、羅馬時期已有類似描述,但直到 1906 年 Alois Alzheimer 系統性報導神經元大量消失與「特殊顆粒狀沉積」後,AD 才被正式命名。
β 類澱粉蛋白(Aβ)發現:1980 年代 Konrad Beyreuther 團隊確認該「特殊物質」為 Aβ,並追溯至 21 號染色體上的 類澱粉前體蛋白(APP)。
類澱粉蛋白假說
核心主張:Aβ 異常聚積為病程起點,啟動發炎、氧化損傷與 過度磷酸化 tau 聚集等連鎖病理。
兩大神經病理標誌:
Aβ 斑塊(plaques)
tau 纏結(tangles)
主要挑戰與反證
無症狀斑塊載體:解剖或影像研究發現,部分智健長者亦有大量 Aβ 斑塊,卻無臨床失智。
抗斑塊療法屢失敗:多數以清除斑塊為目標的臨床試驗未能改善認知(donanemab 為少數例外)。
推論:大型、不溶 Aβ 斑塊可能非真正毒源。
小型可擴散聚集體(oligomers)的興起
毒性新焦點:長度約 100 nm、可在突觸間擴散的 Aβ/tau 小聚集體被認為對突觸功能與細胞訊號具高度毒性,可能比大型斑塊更早驅動病程。
研究門檻:因超出光學繞射極限,需借助單分子螢光與超解析成像技術才能觀測與定量。
治療啟示:鎖定小型聚集體的藥物展現較佳療效,支持其為關鍵致病尺碼。
關鍵科學問題與本文目的
生成機制未明:小聚集體、纖維與斑塊/纏結究竟是「連續光譜」還是「平行途徑」產物?
本文目標:整合最新實驗與單分子研究,探討 小型可擴散聚集體與大型病理產物的形成機制差異,並評估「主動纖維化/封存」對 AD 病程的影響。
圖解fig1
被動連續聚集階段
Monomer → dimer/trimer → oligomer:當 Aβ 或 tau 單體濃度超過溶解度閾值,即依生物物理「二級核化」模型自發聚集,生成 < 100 nm 的小型可擴散聚集體。
此階段毒性最高,直接擾動突觸受體、鈣恆定與基因調控。
主動封存轉換
小聚集體若持續過剩,微膠質會藉吞噬與分泌基質蛋白,將 Aβ 打包成致密斑塊;神經元則透過 caspase 觸發 tau 纖維化,形成纏結。
斑塊/纏結屬「主動防禦產物」,降低游離毒性,但若封存容量飽和,即引爆炎症與神經元耗損。
- 阿茲海默症的病理,始於被動形成的高毒性寡聚體;而我們熟知的斑塊與纏結,則可能是細胞為了應對這些寡聚體而進行的主動防禦反應的結果。
小型可擴散聚集體與斑塊 (Small-diffusible aggregates and plaques)標題
被動路徑:小型可擴散聚集體的形成
物理化學驅動:小型可擴散聚集體(寡聚體)的形成,遵循著基本的生物物理學模型。當蛋白質或胜肽單體的濃度超過其溶解度閾值時,熱力學定律會驅動系統走向聚集。
成核與生長:這個過程通常需要克服一個初始的能量壁壘,即「成核步驟」。一旦最初的聚集核心形成,它便可以透過不斷吸附溶液中的單體來生長,形成不同尺寸與形態的聚集體。
自我複製機制:已形成的聚集體還具有自我複製的能力,例如透過斷裂(fragmentation)產生新的核心,或是在現有聚集體表面進行二級成核(secondary nucleation),從而加速整個聚集過程。
過程本質:總體而言,這種小型聚集體的形成是一個純粹的「被動」物理化學過程,即使在試管中也能發生。在生物體內,雖然有蛋白質品質管控系統會對其進行抑制,但其根本驅動力不變。
主動路徑:斑塊的形成
核心論點:與寡聚體的被動形成相反,由大量 Aβ 纖維構成的斑塊,其形成過程可能是一個由微膠細胞 (microglia) 主動調控的生物學反應。
濃度悖論:一個強力的物理學證據是,大腦細胞外液中 Aβ42 的濃度(約 1–10 nM)遠低於其自發形成纖維所需的臨界聚集濃度(90 nM)。這意味著,自發性的纖維化與斑塊形成在生理條件下難以發生。
細胞生物學證據:
在 AD 小鼠模型中,若用藥物清除微膠細胞,會顯著減少 Aβ 斑塊的形成,但可擴散的寡聚體數量卻保持不變。
微膠細胞表面的 TAM 受體(Axl 和 Mer)在 AD 小鼠模型中會上調。若下調這些受體,則會導致緻密核心斑塊的減少。
結論:這些證據共同表明,斑塊並非由寡聚體被動地持續生長而成,而是由微膠細胞主動介導的結果。
保護性假說及其臨床意涵
假說內容:基於寡聚體具有更高毒性的證據,微膠細胞主動形成斑塊的行為,可能是一種保護性機制。其目的在於將高毒性、可擴散的寡聚體「打包」成體積更大、生物活性更低的物種,以圖恢復大腦內 Aβ 生成與清除的動態平衡。
臨床解釋:
解釋無症狀帶斑塊者:這個假說解釋了為何部分認知功能完好的個體腦中也存在 Aβ 斑塊。在這些個體中,這個「打包」機制可能成功地控制了寡聚體的毒性,從而阻止或延緩了疾病的進展。
解釋時間延遲:它也解釋了為何從斑塊出現到認知功能下降之間,存在著很長的時間差。疾病的進程被延緩,直到這個保護性機制飽和或不堪重負。
疾病進程:只有當寡聚體的生成速度持續超過清除與打包的總和能力時,失控的寡聚體才會引發如 Tau 蛋白病理等次級疾病機制,最終導致認知功能衰退。
小聚集體與斑塊並非單一路徑的「大小連續體」;斑塊更多是由微膠質主動驅動的二次防禦產物。
早期治療策略應聚焦於 抑制小型可擴散聚集體生成或促進其清除,而非僅單純剷除斑塊。
關鍵推論
小聚集體與斑塊並非單一路徑的「大小連續體」;斑塊更多是由微膠質主動驅動的二次防禦產物。
早期治療策略應聚焦於 抑制小型可擴散聚集體生成或促進其清除,而非僅單純剷除斑塊。
阿茲海默病中的 Tau 蛋白聚集體 (Tau aggregates in AD)
纏結的形成:由 Caspase 驅動的主動過程
-
理論推測:在神經元內擁擠的細胞質環境中,Tau 蛋白要自發形成大型纖維結構,面臨著很高的能量壁壘。因此,纏結的形成很可能需要一個「主動」過程的介入。這個過程可能涉及 Caspase 蛋白酶的活化,導致 Tau 蛋白被截斷 (truncation),從而降低了形成纖維(可能是保護性纖維)的能量門檻。
-
活體影像證據:利用小鼠模型進行的活體多光子顯微影像研究,為此提供了強力支持。研究顯示:
-
時序關係:Caspase(特別是 caspase-3 和 -6)的活化,明確地「發生在」纏結形成之前數小時。多光子顯微追蹤顯示,caspase 活化後 ~4 h 內即出現纏結,且隨後 caspase 信號被抑制。
-
因果關聯:有 Caspase 活性的神經元,其形成纏結的機率顯著高於沒有 Caspase 活性的神經元。纏結形成後的神經元 90% 以上可存活至少 28 天,RNA-seq 未見凋亡基因上調,顯示纏結並非直接殺手。
-
活性抑制:一個值得注意的現象是,在纏結形成「之後」,神經元內的 Caspase 活性反而會減弱。
-
纏結的保護性角色與神經元存活
-
神經元存活證據:多項研究指出,帶有纏結的神經元不僅能夠存活,還能維持其在神經迴路中的功能。對這些細胞的轉錄組分析也顯示,與細胞死亡相關的基因並無顯著變化。這直接挑戰了「纏結是細胞死亡標誌」的傳統觀念。
-
「雙重效益」假說:當細胞內有毒的可溶性 Tau 寡聚體累積到一定程度,會觸發 Caspase 活化,引導細胞走向凋亡。此時,形成纏結可能是一種自救機制,它能帶來雙重好處。
-
清除毒物:將有毒的可溶性 Tau 寡聚體打包成相對惰性的纏結,降低其在細胞質中的濃度。
-
抑制凋亡:纏結的形成能反過來抑制 Caspase 的活性,從而使神經元免於死亡。
-
-
臨床相關性:這個假說也解釋了為何纏結的出現與神經功能缺損高度相關——因為形成纏結是一個高風險下的「成功救援」事件,在大多數情況下,Caspase 的活化可能直接就導致了細胞死亡,無法成功形成纏結。
可溶性 Tau 聚集體:真正的毒性來源
-
動物模型證據:
-
越來越多的證據將矛頭指向可溶性 Tau 聚集體。新開發的 tauopathy 小鼠模型顯示,即使沒有形成纖維狀纏結,僅僅是非纖維性 Tau 聚集體的累積,就足以造成突觸喪失和行為異常。
-
小聚集體可破壞突觸鈣通道(CaV2.3)、抑制複雜放電,導致突觸失能與行為異常;小鼠 tauopathy 模型中,高分子量可溶 tau 是主要病理驅動因子。
-
-
電生理學證據:對 AD 小鼠模型的海馬迴神經元進行的電生理記錄顯示,神經元放電模式的異常,發生在纏結形成之前,並且主要是由可溶性、高分子量的 Tau 物種所引起。
-
人腦樣本證據:對 AD 患者腦組織的分析發現,不溶性 Tau 的含量與患者的存活年齡呈「正相關」(即不溶性 Tau 越多,存活越久)。相比之下,特定位點磷酸化(S396)的可溶性 Tau 則與存活年齡呈「負相關」。這強烈暗示了可溶性與不溶性 Tau 在病理中扮演著截然不同的角色。
-
「幽靈纏結」的啟示:因神經元死亡而遺留在細胞外的「幽靈纏結」(ghost tangles),在 AD 患者腦中僅佔所有纏結的約 0.7%。這進一步證實,在疾病的終末期,絕大多數帶有纏結的神經元仍然是存活的。
多階段動態平衡假說 (The multi-stage dynamic equilibria hypothesis)
第一階段:Aβ 平衡的破壞與初級代償
Aβ 的生理角色:研究顯示,Aβ 在中樞神經系統中可能扮演著生理性的角色,例如在發炎等挑戰下作為一種「損傷反應元件」。在正常情況下,由發炎刺激(如 TNF-α)產生的少量 Aβ 寡聚體會被有效清除,而不會觸發下游的 Tau 病理。
平衡的破壞:然而,當此平衡因持續的刺激或遺傳因素(如家族性 AD 突變)而被進一步打破時,可擴散的 Aβ 寡聚體便會開始大量累積。
初級代償:斑塊的形成:此時,微膠細胞會被活化,啟動一個初級的、具有保護性質的代償機制——主動將這些高毒性的寡聚體「打包」成相對惰性的緻密核心斑塊。
斑塊的雙重角色:需要澄清的是,此假說並非認為斑塊是完全無害的。斑塊本身確實會引發局部的發炎反應。然而,相較於能夠自由擴散、生物活性極高的小型寡聚體,由大量寡聚體形成的一個大型、固定的斑塊,其相對毒性較低。
臨床解釋:這個「二級安全機制」解釋了為何認知功能完好的個體腦中也存在 Aβ 斑塊。在這些案例中,形成斑塊的機制成功地降低了寡聚體的濃度,從而恢復了平衡,阻止或延緩了 Tau 病理等下游事件的發生。
第二階段:Tau 平衡的破壞與二級代償
代償失靈與病理級聯:如果寡聚體的生成速度持續超過清除與打包的總和能力,這個初級代償機制便會不堪重負。失控的 Aβ 寡聚體會透過氧化壓力、發炎等多種途徑,最終引發下游的 Tau 病理。
二級代償:纏結的形成:同樣的平衡邏輯也適用於 Tau 蛋白。面對具有突觸毒性和神經毒性的過度磷酸化 Tau 聚集體,神經元會啟動二級代償機制,透過 Caspase 活化等過程,將其「鈍化」並打包成神經纖維纏結。
系統崩潰與功能喪失:一旦這個最後的防線也被突破,生成與清除的平衡徹底崩潰,突觸與神經元毒性便會全面爆發,最終導致與 AD 相關的認知和行為功能障礙。
結論、局限與未來方向 (Conclusion, limitations, and future directions)
動態平衡失衡是核心病理
AD 的慢性進展源自 小型可擴散 Aβ/tau 聚集體 的產生-清除失衡;斑塊與纏結屬於微膠質與神經元為降低毒性而啟動的 二次封存機制。
毒性主角重新定位
質量龐大的斑塊/纏結 ≠ 主要殺手;高生物活性的 diffusible aggregates 才驅動發炎、突觸失能與基因調控異常。
診斷指標需前移
應開發可檢測 nano-oligomer 的血液/CSF 或影像工具,而非僅追蹤 PET 斑塊負荷。
生理層面存在微量聚集體
小聚集體並非病理專屬;健康腦亦持續生成並清除,失衡才導向神經退化。
重新詮釋現有藥物:
Lecanemab:其成功可被視為此假說的有力支持,因為它主要靶向的正是疾病早期的、小型的 Aβ 聚集體。
Donanemab:該藥物主要清除斑塊,看似與假說矛盾。但其療效可被解釋為:1) 間接清除了斑塊周圍高濃度的寡聚體;2) 移除斑塊這個巨大的 Aβ 庫存,改變了化學平衡,促使更多寡聚體解離。
未來藥物開發的方向:
靶點轉移:一個重要的結論是,不溶性的斑塊和纏結並非理想的治療監控指標或靶點。下一代療法的開發,應著重於建立能評估藥物與小型寡聚體結合能力的生物測定法 (bioassay)。
精準靶向:應避免無差別地靶向 Aβ 或 Tau。例如,若纏結與截斷的 Tau 蛋白具有保護性,那麼靶向它們的療法可能反而有害。未來的策略應是開發能特異性識別並作用於真正致病的聚集體亞型的藥物。
- 對診斷工具的未來要求
- 目前主流的 PET 影像與多數體液生物標記,主要偵測的是大型、不溶性的聚集體。然而,為了配合早期治療(如 Lecanemab),我們迫切需要開發能夠特異性偵測早期、微量的可溶性聚集體的新型超靈敏診斷工具。
對既往研究的重新評估 (Re-evaluation of Previous Experimental Findings)
若「多階段動態平衡假說」獲得進一步驗證,將會要求學界對過去數十年來積累的大量實驗發現,進行一次根本性的重新評估。
種子實驗 (Seeding Experiments) 的重新解讀
現行方法:許多細胞培養的「種子」實驗,是使用從 AD 大腦中提取的、不溶於 sarkosyl 的組分(即大型、不溶性的纖維)作為「種子」來誘導聚集。
新觀點:在此假說下,這些被當作「種子」的不溶性聚集體,其本質可能是保護性的,而非毒性的。
現象解釋:這個觀點也能解釋為何在此類種子實驗中,通常觀察不到顯著的細胞死亡現象。
動物模型研究 (Animal Model Studies) 的焦點轉移
現行文獻的局限:目前的動物模型研究,大多將觀察到的毒性機制與可見的不溶性斑塊和纏結的數量進行關聯。
潛在的真正元兇:根據此假說,真正驅動病理的,可能是那些與大型聚集體同時形成、卻因技術限制而「不可見」的小型可擴散寡聚體。
未來方向:因此,未來的動物模型研究,必須採用能夠特異性偵測這些小型寡聚體的先進方法,才能準確地將病理機制與真正的致病因子連結起來。
冷凍電鏡結構 (Cryo-EM Structures) 評估
現行研究的重點:近年來,利用冷凍電子顯微鏡 (Cryo-EM) 解析不溶性纖維的精美原子結構,是結構生物學領域的一大熱點。
新觀點:此假說提出,這些被解析的結構,可能代表的是已被鈍化的、處於終末期的保護性物種,而非具有活性的致病毒物。
治療策略的警示:若以此類結構為基礎來設計免疫療法,其目標可能是錯誤的。靶向這些可能具有保護作用的結構,不僅可能無效,甚至可能對患者產生不利的影響。
想法: 此假說的進展
Aβ/Tau 核心想法不變
此研究並未顛覆類澱粉蛋白與 Tau 病理假說的時序基礎,只是將「毒性病理焦點」從沉積的蛋白質轉移至 可溶可擴散的寡聚體。
被動 vs. 主動二元聚集機制
被動:Aβ/Tau 寡聚體生成受熱力學驅動;
主動:斑塊與纏結形成需細胞介入
此模型將純生物物理過程與細胞反應清楚區分,較線性級聯理論具有更高解釋力。
對目前病理和藥物治療的新解釋
認知正常長者亦可見高斑塊負荷代表封存機制仍有效,可溶性寡聚體減少。
大多清除類澱粉斑塊的藥物無臨床效益因為鎖定的是相對次要或已封存的構造。
對現行抗類澱粉藥物治療成效的詮釋
Lecanemab — 直接中和寡聚體,降低突觸毒性。
Donanemab — 清除斑塊並間接移除周邊寡聚體,或透過質量作用定律下調寡聚體濃度;說明斑塊仍具可被利用的治療價值,但並非最終治療靶點。
但潛在邏輯弱點: 若斑塊具保護性,donanemab 的臨床受益則需更多實驗證據支持「伴隨減少寡聚體」或「平衡位移」機制,否則會削弱「斑塊應保留」之論述絕對性。
需在臨床的長期追蹤研究中,開發可測量血液/CSF oligomer 的技術或 <開發出可顯示寡聚體的PET 示踪劑,來證實上理論。
在治療的發展上,藥物篩選須納入「對寡聚體有相對親和力」的特性;anti-tau是否更應聚焦可容性的tau,而非清除纏結。
個人化治療時間窗:捕捉 Aβ 和tau 開始失衡 或之黃金治療窗口。
| 平衡狀態 | 病理標誌 | 功能狀態 | 狀態解釋 |
|---|---|---|---|
| 平衡維持 | 少量 Aβ/tau 聚集體 | 正常 | 生理炎症後恢復 |
| 第一次失衡 | 小 Aβ↑ | 潛伏 | 老化初期 |
| 封存成功 | 高斑塊、低小 Aβ | 無症但斑塊陽性 | 認知正常帶斑塊長者 |
| 封存飽和 | 小 Aβ↑、微膠質活化 | 前臨床/SCD/ MCI | Plaque+、Tau− 早期個案 |
| 二次封存 | 高纏結、低可溶 tau | MCI/ mild dementia | NFT 多但神經元存活 |
| 完全失衡 | 可溶 tau↑[Synapse] | AD dementia | tau oligomer 關鍵毒性神經細胞死亡 |
重要問答
Q1:傳統的類澱粉蛋白假說將 Aβ 斑塊與 Tau 纏結視為核心病理,本文的核心論點如何對此提出修正?
A:此文的核心論點在於對 Aβ 斑塊與 Tau 蛋白纏結的「角色」進行了重新定義。傳統觀點將這兩者視為驅動神經毒性的主要因子,但本文整合近年證據提出,它們更可能是一種由細胞主動調控的「保護性機制」的最終產物 。該假說認為,真正引發病理級聯反應的,是體積更小、可溶且具有高度生物活性的寡聚體 (oligomers) 。而大型、不溶性的斑塊與纏結,則是大腦中的小膠質細胞與神經元為了隔離和鈍化這些高毒性寡聚體,所進行的一種積極的應對措施 。
Q2:鑑於腦中 Aβ 單體濃度遠低於自發聚集的門檻,本文如何解釋大規模 Aβ 斑塊形成的?
A:此文獻透過整合生物物理學與細胞生物學的證據,對此提出了合理解釋。關鍵在於,斑塊的形成並非一個被動的物理化學過程,而是一個由小膠質細胞 (microglia) 主導的「主動生物過程」 。腦中 Aβ42 的生理濃度 (約 1-10 nM) 確實遠低於其自發形成纖維所需的臨界聚集濃度 (90 nM) 。因此,文章推論,是小膠質細胞辨識並吞噬了早期的 Aβ 聚集體,並將其主動地「壓實」成結構更緻密、生物活性更低的核心斑塊 。這個由免疫細胞介導的過程,解釋了為何在單體濃度不足的情況下,依然能觀察到大規模的斑塊沉積。
Q3:神經纖維纏結 (NFTs) 的數量與認知能力下降高度相關,但本文對其在細胞層面的角色提出了什麼不同的解釋?
A:儘管在宏觀上 NFTs 的數量是衡量疾病嚴重程度的指標 ,本文整合的微觀證據卻挑戰了「纏結直接導致細胞死亡」的傳統觀點。研究指出,NFTs 的形成並非細胞功能衰敗的終點,而可能是一個在極端壓力下被觸發的、具有保護潛力的細胞反應 。關鍵的活體影像證據顯示,一種促進細胞凋亡的 Caspase 蛋白酶,其活性在 NFTs 形成「之前」會顯著升高,但在纏結形成「之後」反而會減弱 。更重要的是,研究證實帶有纏結的神經元能夠繼續存活並維持其在迴路中的功能 。因此,本文提出的新觀點是:形成纏結可能是神經元為應對毒性 Tau 寡聚體累積、並抑制 Caspase 介導的死亡路徑而採取的一種應急機制 。
Q4:如果斑塊和纏結並非病理的根本原因,這套「動態平衡假說」認為阿茲海默症的觸發點是什麼?
A:此假說將阿茲海默症的觸發點,從聚集體的「存在」轉向了蛋白質穩態的「失衡」 。其核心觀點是,AD 的啟動並非因為 Aβ 或 Tau 蛋白的產生,而是因為其「清除效率的衰退」導致了生成與清除之間的動態平衡被打破 。當這個平衡向生成端傾斜,高毒性的可溶性寡聚體便開始淨增加,超出系統的負荷能力 。此時,細胞啟動了形成斑塊和纏結等次級、代償性的應對機制 。因此,疾病的根本驅動因素是蛋白質清除體系的失效,而非聚集體本身。
Q5:此假說如何為 Lecanemab 與 Donanemab 這兩種 Aβ 抗體藥物看似不同的作用機制提供統一的解釋框架?
A:該假說為這兩種藥物的臨床效果提供了一個統一的生物學解釋。
Lecanemab 的作用機制與假說高度契合,它主要靶向並清除被認為毒性最強的可溶性 Aβ 寡聚體與原纖維 (protofibrils)
而 Donanemab 雖然主要靶向已沉積的斑塊,但其療效可被解釋為兩種間接效應的結果:其一,可能清除了斑塊周圍高濃度的寡聚體;其二,清除了斑塊這個巨大的「寡聚體儲存庫」,進而降低其整體濃度 。因此,儘管直接靶點不同,兩種療法可能都透過降低可溶性寡聚體的有效濃度而發揮作用。
Q6:若此假說成立,對於阿茲海默症未來的診斷策略與研究方向有何意義?
A:這將引導領域的資源和研究方向發生重大轉移。未來的重點必須從觀測相對晚期、大型的不溶性聚集體,轉向偵測和量化早期、微量且高毒性的可溶性寡聚體 。
在診斷策略上,這意味著需大力發展能夠精準測量血液或腦脊液中寡聚體水平的超靈敏生物標記 。
研究方向上,則需要設計更能區分不同聚集形態生物學效應的實驗模型,並對過去基於不溶性聚集體的研究結論,在此新框架下進行批判性的重新評估